

Blog
Neurofibromatosis: associated tumours of childhood in the molecular era
Monday September 6, 2021
Neurofibromatosis (NF) is a multi-systemic genetic disorder characterised by benign indolent neurofibromas in subcutaneous and deep locations but is also associated with other tumours. Neurofibromatosis 1 (NF1) is its most common form (96 per cent), followed by Neurofibromatosis 2 (NF2) and Schwannomatosis (NF3). Although all types affect the nerve sheath, they are clinically and genetically unique entities that merit individualised management strategies.
This review focuses on benign and malignant tumours seen in children and young adults with NF and highlights the growing impact of a precision oncology approach on conventional clinical care.
Genetic pathology
The NF1 gene is located on chromosome 17q11.2, with alterations leading to reduction or loss of function of its protein neurofibromin, 50 per cent of which occur in the germline. The resulting downstream dysregulation of the mitogen-activated protein kinase (MAPK) signalling pathway stimulates cellular proliferation, tumour growth
and survival. Subcutaneous and plexiform neurofibromas are the most common benign NF1-mutant tumours but 10 per cent can potentially transform into malignant peripheral nerve sheath tumours (MPNST). In addition
to NF1, aberrations in SUZ12, EED, TP53 and CDKN2A frequently cooperate to drive their aggression. Alongside NF1, recurrent BRAF and MEK activation contribute to upregulating the same pathway in intracranial low-grade gliomas (LGG) in the optic pathway (OPG) and brainstem, as well as in high-grade gliomas.
The NF2 gene resides on chromosome 22q12.1, encoding merlin or schwannomin. Germline alterations in this
gene lead to vestibular schwannomas (VS) (or acoustic neuromas) in 90 per cent of cases. Supplemental lesions in CHEK2, POLE, ATM and BRAF are observed in 50 per cent of NF2-driven high-grade meningiomas, while NF2- wild type meningiomas can demonstrate TERT promoter and TP53 mutations. NF2 alteration can lead to cervical or cervico-medullary spinal ependymomas.
Schwannomatosis (NF3) is associated with germline mutations in SMARCB1 (chromosome 22q.23.79) and LZTR1 (chromosome22q.11.21), in turn upregulating the MAPK pathway, resulting in deeper non-cancerous neurofibromata.
Mosaic or segmental NF can result in all the above forms. While all NF sub-groups display autosomal dominant inheritance, NF1 is nearly completely penetrant while NF2 and NF3 are variably penetrant.
Tumour manifestations
Clinical features depend on location, with emerging genotype-phenotype correlations. Typical neurological presentations include visual impairment and obstructive hydrocephalus, while neurofibromatous pressure effects can cause significant pain, disfigurement and dysfunction.
VNs can result in hearing impairment and tinnitus, but any or all of these can be debilitating for many and life- threatening for some patients. Although overall survival is >90 per cent, these tumours carry high morbidity burden, i.e. visual, endocrine, neurological, cognitive function and second malignancy.
Diagnosis
NF is essentially a clinical diagnosis, guided by well-established, internationally accepted diagnostic criteria. Confirmatory genomic sequencing on tissue, blood or saliva is available but reserved for diagnostic dilemma (mosaicism, segmental NF or overlap with other genetic conditions) or pre-natal genetic diagnosis (PGD) in asymptomatic individuals with family history of NF. Linkage analysis could prove useful if mutational approaches are inconclusive. However, genetic testing cannot currently predict the severity of any form of neurofibromatosis.
Management
NF is unfortunately not curable. However, most manifestations are managed conservatively, often with observation alone.
Invasive intervention is predicated on tumour location and symptoms. Vision preservation is the primary indication for conventional chemotherapy (e.g. vincristine/carboplatin/ vinblastine) in optic pathway gliomas (OPGs). Anti-angiogenic agents like bevacizumab have shown benefit against OPG and VS.
Maximal safe surgical resection is still the mainstay of management for MPNST, ependymoma, meningioma and symptomatic neurofibroma but bears significant risk of surgical morbidity.
While radiotherapy is not routinely recommended in NF patients due to germline cancer predisposition and risk of vasculopathy (MoyaMoya disease), focal radiotherapy for symptomatic surgically inaccessible neurofibromas and recurrent gliomas should be an informed individualised decision.
The conspicuous MAPK pathway activation in NF tumours makes molecularly selective targeting an increasingly attractive option. BRAF (dabrafinib/vemurafenib) and MEK (trametinib, selumetinib) inhibitors have demonstrated promising outcomes for symptomatic, progressive or refractory tumours. Ongoing and future trials will hopefully establish long-term efficacy and safety against conventional strategies and potentiate promotion for use in frontline therapies. Day101, a novel pan-RAF inhibitor is currently undergoing clinical evaluation for progressive low-grade gliomas and may help bring better balance of benefit and toxicity. Recent studies indicate the potential benefit of predictive genomic biomarkers in early identification of high-risk patients for VS-associated hearing loss, that could mitigate auditory morbidity.
Lifelong multi-disciplinary surveillance is recommended for patients with pathogenic germline variants, with early referral to the Clinical Genetics team for family counselling.
The figure above illustrates the differential spectrum of NF1-associated malignancies.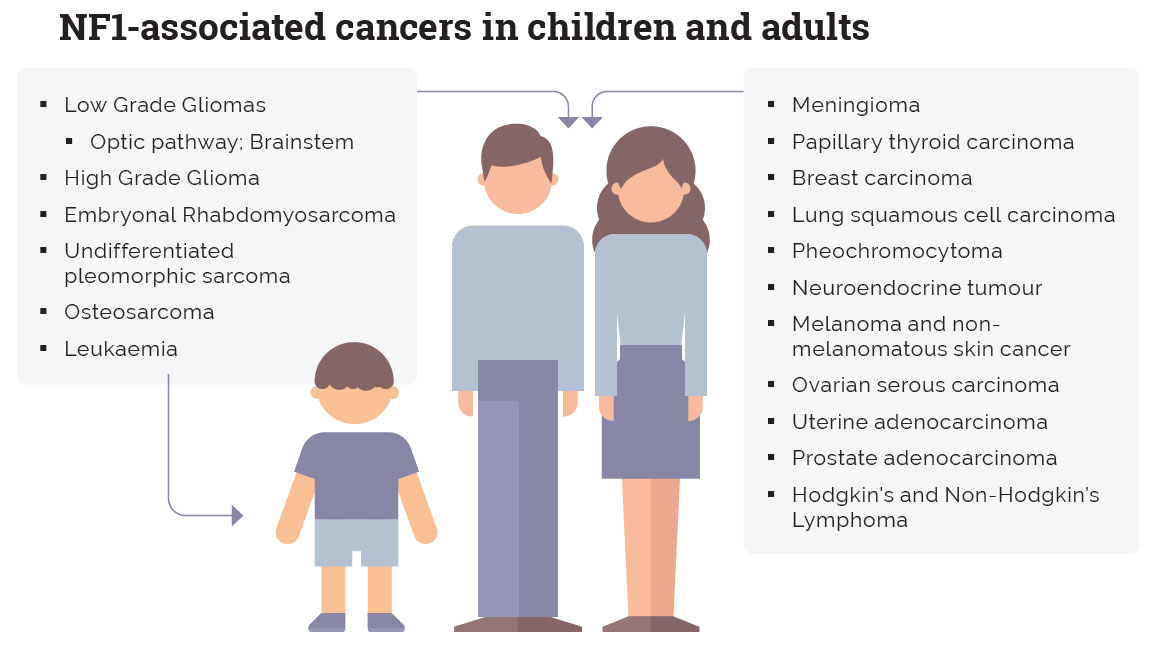
The figure above illustrates the differential spectrum of NF1-associated malignancies.
Future perspectives
The exponential development of genomic technology, consistently improving experience in deep molecular profiling and clinical translational ability, has transformed our outlook to treating these mostly slow-growing but morbidity-heavy tumours. This has contributed to our enhanced understanding of the biological underpinnings of this complex disease, providing new insights into the NF genetic landscape and uncovering actionable genetic faults exploitable for therapeutic benefit in patients otherwise lacking treatment recourse.
This personalised approach is an emerging yet vital tool to augment patient survival and reduce treatment effects, in this potentially severe, debilitating and life-threatening genetic condition.
Research funding welcome
To this end, a $7 million Federal Government-fronted Medical Research Future Fund (MRFF) Neurofibromatosis Research Grant Opportunity was established to support ground-breaking research to develop a better understanding on the variability within these complex conditions and new interventions to improve outcomes for patients with NF. Recently the MRFF announced that $4.6 million in grants was awarded to four Australian teams for performing research into the genetics of cutaneous neurofibromas, blood sample-based biomarker discovery for MPNST, trialling listening devices for NF1-associated auditory processing disorder and to define and target function areas based on the structure and genetics of NF1.
The Children’s Tumour Foundation of Australia (CTF) is the peak national body supporting the NF community. It provides individualised and systemic advocacy and support, as well as community programs, educational resources, clinical and research support and funding across the country. The organisation’s advocacy efforts delivered the recent MRFF Neurofibromatosis grant round and it is committed to continued engagement and support such as these and other projects.
 Dr Sumanth Nagabushan
Dr Sumanth Nagabushan
MBBS MRCPCH FRACP
Dr Sumanth Nagabushan is a paediatric oncologist with subspecialty training in Neuro-Oncology and precision medicine. He currently works at Perth Children’s Hospital.
Acknowledgement: I would like to thank Dr Hetal Dholaria, paediatric oncologist, Perth Children’s Hospital for her review of the article and the Children’s Tumour Foundation Australia for its support.